Glow 0.3.0 Introduces Several New Large-Scale Genomic Analysis Features
Glow 0.3.0 was released on February 21, 2020, improving Glow’s power and ease of use in performing large-scale genomic analysis. In this blog, we highlight features and improvements introduced in this release.
Python and Scala APIs for Glow SQL functions
In this release, Python and Scala APIs were introduced for all Glow SQL functions, similar to what is available for Spark SQL functions. In addition to improved simplicity, this provides enhanced compile-time safety. The SQL functions and their Python and Scala clients are generated from the same source so any new functionality in the future will always appear in all three languages. Please refer to PySpark Functions for more information on Python APIs for these functions. As an example, the usage of such Python and Scala APIs for the function normalize_variant is presented at the end of next section.
Improved variant normalization
The variant normalizer received a major improvement in this release. It still behaves like bcftools norm and vt normalize, but is about 2.5x faster and has a more flexible API. Moreover, the new normalizer is implemented as a function in addition to a transformer.
normalize_variants transformer: The improved transformer preserves the columns of the input DataFrame, adds the normalization status to the DataFrame, and has the option of adding the normalization results (including the normalized coordinates and alleles) to the DataFrame as a new column. As an example, assume we read the original_variants_df DataFrame shown in Fig. 12 by issuing the following command:
original_variants_df = spark.read \
.format("vcf") \
.option("includeSampleIds", False) \
.load("/databricks-datasets/genomics/call-sets")
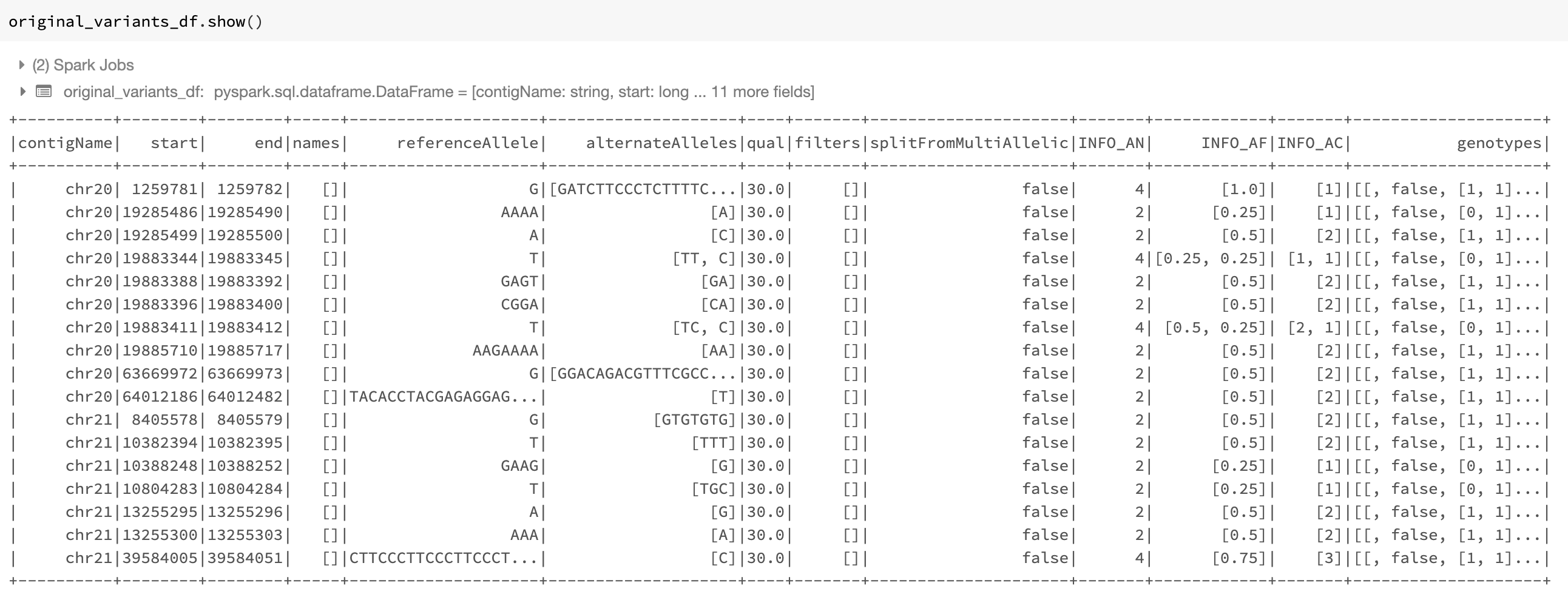
Fig. 12 The variant DataFrame original_variants_df
The improved normalizer transformer can be applied on this DataFrame using the following command similar to the previous version of the normalizer:
import glow
normalized_variants_df = glow.transform("normalize_variants", \
original_variants_df, \
reference_genome_path="/mnt/dbnucleus/dbgenomics/grch38/data/GRCh38_full_analysis_set_plus_decoy_hla.fa" \
)
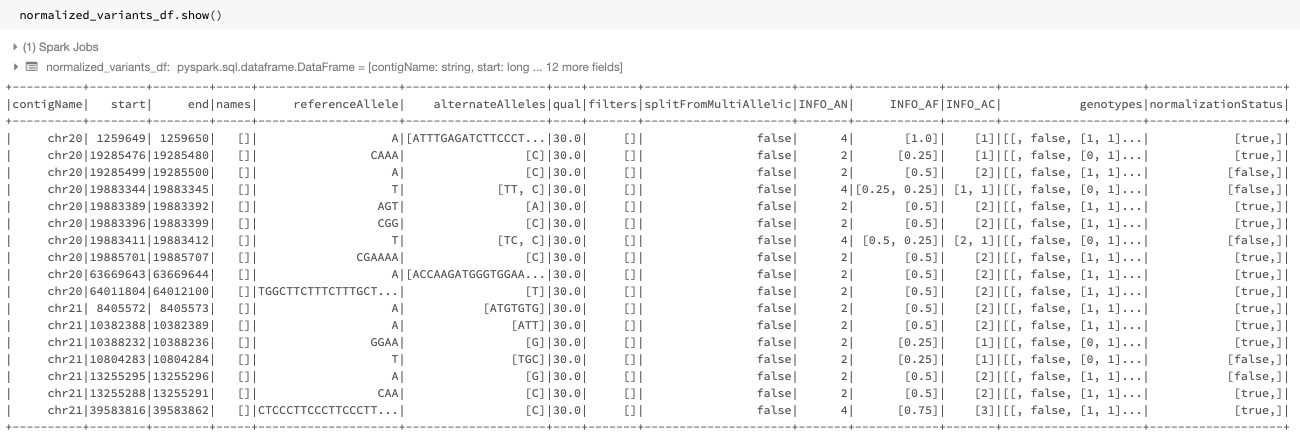
Fig. 13 The normalized DataFrame normalized_variants_df
The output DataFrame of this improved transformer looks like Fig. 13. The start, end, referenceAllele, and alternateAlleles fields are updated with the normalized values and a normalizationStatus column is added to the DataFrame. This column contains a changed subfield indicating whether normalization changed the variant and an errorMessage subfield containing the error message in case of an error.
The newly introduced replace_columns option can be used to add the normalization results as a new column to the DataFrame instead of replacing the original start, end, referenceAllele, and alternateAlleles fields. This can be done as follows:
import glow
normalized_variants_df = glow.transform("normalize_variants",\
original_variants_df, \
replace_columns="False", \
reference_genome_path="/mnt/dbnucleus/dbgenomics/grch38/data/GRCh38_full_analysis_set_plus_decoy_hla.fa" \
)
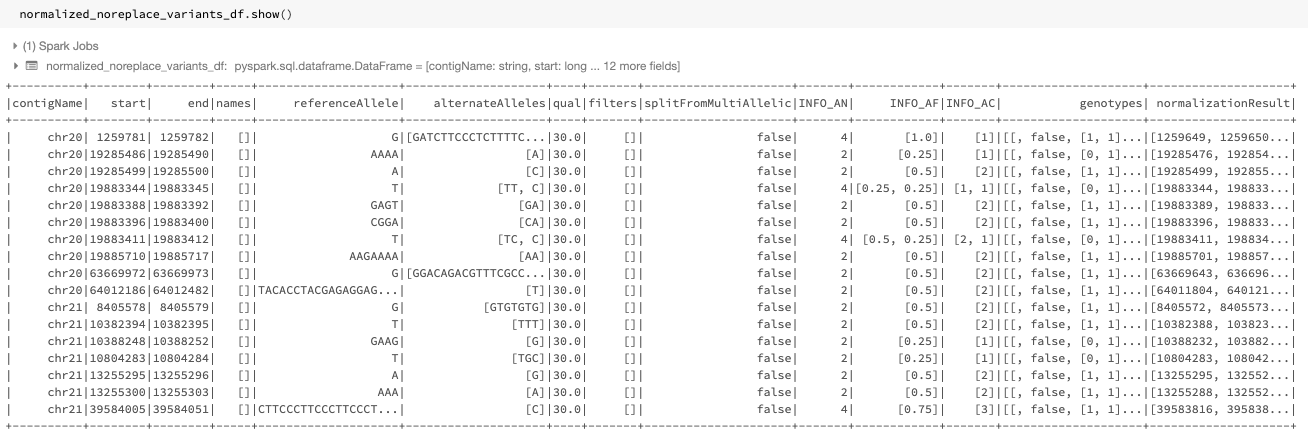
Fig. 14 The normalized DataFrame normalized_noreplace_variants_df with normalization results added as a new column
The resulting DataFrame will be as shown in Fig. 14, where a normalizationResults column containing the normalized start, end, referenceAllele, alternateAlleles, and normalizationStatus subfields is added to the DataFrame.
We also note that since the multiallelic variant splitter is implemented as a separate transformer in this release (see below), the mode option of the normalize_variants transformer is deprecated. Refer to Variant Normalization for more details on the normalize_variants transformer.
normalize_variant function: As mentioned above, in this release, variant normalization can also be performed using the newly introduced normalize_variant SQL expression function as shown below:
from pyspark.sql.functions import expr
function_normalized_variants_df = original_variants_df.withColumn( \
"normalizationResult", \
expr("normalize_variant(contigName, start, end, referenceAllele, alternateAlleles, '/mnt/dbnucleus/dbgenomics/grch38/data/GRCh38_full_analysis_set_plus_decoy_hla.fa')") \
)
As discussed in the previous section, this SQL expression function, like any other in Glow, now has Python and Scala APIs as well. Therefore, the same can be done in Python as follows:
from glow.functions import normalize_variant
function_normalized_variants_df = original_variants_df.withColumn( \
"normalizationResult", \
normalize_variant( \
"contigName", \
"start", \
"end", \
"referenceAllele", \
"alternateAlleles", \
"/mnt/dbnucleus/dbgenomics/grch38/data/GRCh38_full_analysis_set_plus_decoy_hla.fa" \
) \
)
and in Scala as well, assuming original_variant_df is defined in Scala:
import io.projectglow.functions.normalize_variant
import org.apache.spark.sql.functions.col
val function_normalized_variants_df = original_variants_df.withColumn(
"normalizationResult",
normalize_variant(
col("contigName"),
col("start"),
col("end"),
col("referenceAllele"),
col("alternateAlleles"),
"/mnt/dbnucleus/dbgenomics/grch38/data/GRCh38_full_analysis_set_plus_decoy_hla.fa"
)
)
The result of any of the above commands will be the same as Fig. 14.
A new transformer to split multiallelic variants
This release also introduced a new DataFrame transformer, called split_multiallelics, to split multiallelic variants into biallelic ones with a behavior similar to vt decompose with -s option. This behavior is significantly more powerful than the behavior of the previous version’s splitter which behaved like GATK’s LeftAlignAndTrimVariants with --split-multi-allelics. In particular, the array-type INFO and genotype fields with elements corresponding to reference and alternate alleles are split “smart”ly (see -s option of vt decompose) into biallelic rows. So are the array-type genotype fields with elements sorted in colex order of genotype calles, e.g., the GL, PL, and GP fields in the VCF format. Moreover, an OLD_MULTIALLELIC INFO field is added to the DataFrame to store the original multiallelic form of the split variants.
The following is an example of using the split_multiallelic transformer on the original_variants_df. The resulting DataFrame is as in Fig. 15.
import glow
split_variants_df = glow.transform("split_multiallelics", original_variants_df)
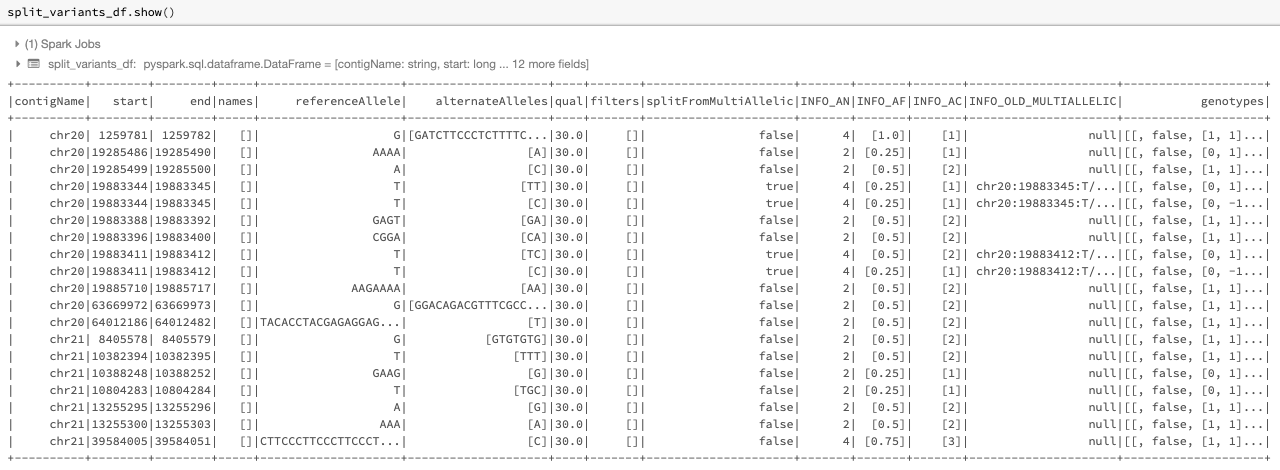
Fig. 15 The split DataFrame split_variants_df
Please note that the new splitter is implemented as a separate transformer from the normalize_variants transformer. Previously, splitting could only be done as one of the operation modes of the normalize_variants transformer using the now-deprecated mode option.
Please refer to the documentation of the split_multiallelics transformer for complete details on the bahavior of this new transformer.
Parsing of Annotation Fields
The VCF reader and pipe transformer now parse variant annotations from tools such as SnpEff and VEP. This flattens the ANN and CSQ INFO fields, simplifying and accelerating queries on annotations. See the following query and its result in Fig. 16 for an example.
from pyspark.sql.functions import expr
variants_df = spark.read\
.format("vcf")\
.load("dbfs:/databricks-datasets/genomics/vcfs/loftee.vcf")
annotated_variants_df = original_variants_df.withColumn( \
"Exploded_INFO_CSQ", \
expr("explode(INFO_CSQ)") \
) \
.selectExpr("contigName", \
"start", \
"end", \
"referenceAllele", \
"alternateAlleles", \
"expand_struct(Exploded_INFO_CSQ)", \
"genotypes" \
)
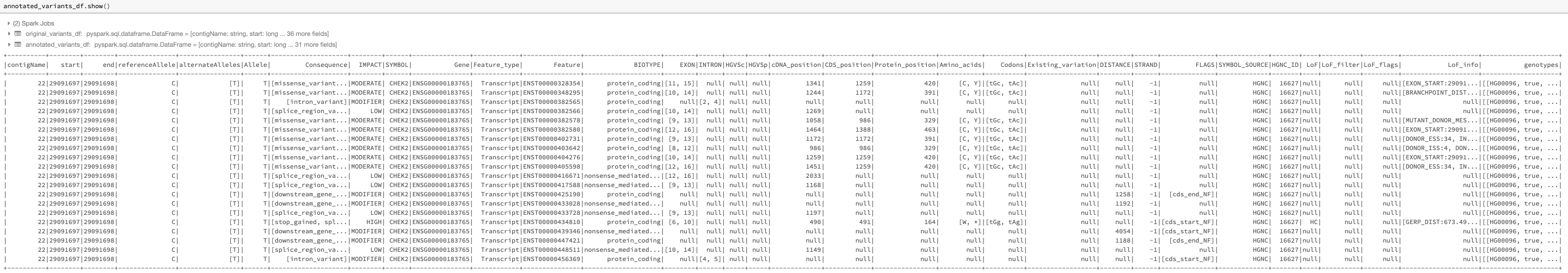
Fig. 16 The annotated DataFrame annotated_variants_df with expanded subfields of the exploded INFO_CSQ
Other Improvements
Glow 0.3.0 also includes optimized implementations of the linear and logistic regression functions, resulting in ~50% performance improvements. See the documentation at Linear regression and Logistic regression.
Furthermore, the new release supports Scala 2.12 in addition to Scala 2.11. The maven artifacts for both Scala versions are available on Maven Central.
Try It!
Try Glow 0.3.0 and its new features here.