Introducing GloWGR: An industrial-scale, ultra-fast and sensitive method for genetic association studies
Authors: Leland Barnard, Henry Davidge, Karen Feng, Joelle Mbatchou, Boris Boutkov, Kiavash Kianfar, Lukas Habegger, Jonathan Marchini, Jeffrey Reid, Evan Maxwell, Frank Austin Nothaft
June 22, 2020
The industry partnership between Regeneron and Databricks is enabling innovations in genomics data processing and analysis. Today, we announce that we are making a new whole genome regression method available to the open source bioinformatics community as part of Project Glow.
Large cohorts of individuals with paired clinical and genome sequence data enable unprecedented insight into human disease biology. Population studies such as the UK Biobank, Genomics England, or Genome Asia 100k datasets are driving a need for innovation in methods for working with genetic data. These methods include genome wide association studies (GWAS), which enrich our understanding of the genetic architecture of the disease and are used in cutting-edge industrial applications, such as identifying therapeutic targets for drug development. However, these datasets pose novel statistical and engineering challenges. The statistical challenges have been addressed by tools such as SAIGE and Bolt-LMM, but they are difficult to set up and prohibitively slow to run on biobank-scale datasets.
In a typical GWAS, a single phenotype such as cholesterol levels or diabetes diagnosis status is tested for statistical association with millions of genetic variants across the genome. Sophisticated mixed model and whole genome regression-based approaches have been developed to control for relatedness and population structure inherent to large genetic study populations when testing for genetic associations; several methods such as BOLT-LMM, SAIGE, and fastGWA use a technique called whole genome regression to sensitively analyze a single phenotype in biobank-scale projects. However, deeply phenotyped biobank-scale projects can require tens of thousands of separate GWASs to analyze the full spectrum of clinical variables, and current tools are still prohibitively expensive to run at scale. In order to address the challenge of efficiently analyzing such datasets, the Regeneron Genetics Center has just developed a new approach for the whole-genome regression method that enables running GWAS across upwards of hundreds of phenotypes simultaneously. This exciting new tool provides the same superior test power as current state-of-the-art methods at a small fraction of the computational cost.
This new whole genome regression (WGR) approach recasts the whole genome regression problem to an ensemble model of many small, genetic region-specific models. This method is described in a preprint released today, and implemented in the C++ tool regenie. As part of the collaboration between the Regeneron Genetics Center and Databricks on the open source Project Glow, we are excited to announce GloWGR, a lightning-fast and highly scalable distributed implementation of this WGR algorithm, designed from the ground up with Apache Spark and integrated with other Glow functionality. With GloWGR, performing WGR analyses on dozens of phenotypes can be accomplished simultaneously in a matter of minutes, a task that would require hundreds or thousands of hours with existing state-of-the-art tools. Moreover, GloWGR distributes along both the sample and genetic variant matrix dimensions, allowing for linear scaling and a high degree of data and task parallelism. GloWGR plugs seamlessly into any existing GWAS workflow, providing an immediate boost to association detection power at a negligible computational cost.
Achieving High Accuracy and Efficiency with Whole-Genome Regression
This whole genome regression tool has a number of virtues. First, it is more efficient: as implemented in the single node, open-source regenie tool, whole genome regression is orders of magnitude faster than either SAIGE, Bolt-LMM, or fastGWA, while producing equivalent results (Figure 1). Second, it is straightforward to parallelize: in the next section, we describe how we implemented whole genome regression using Apache Spark™ in the open-source Project Glow.
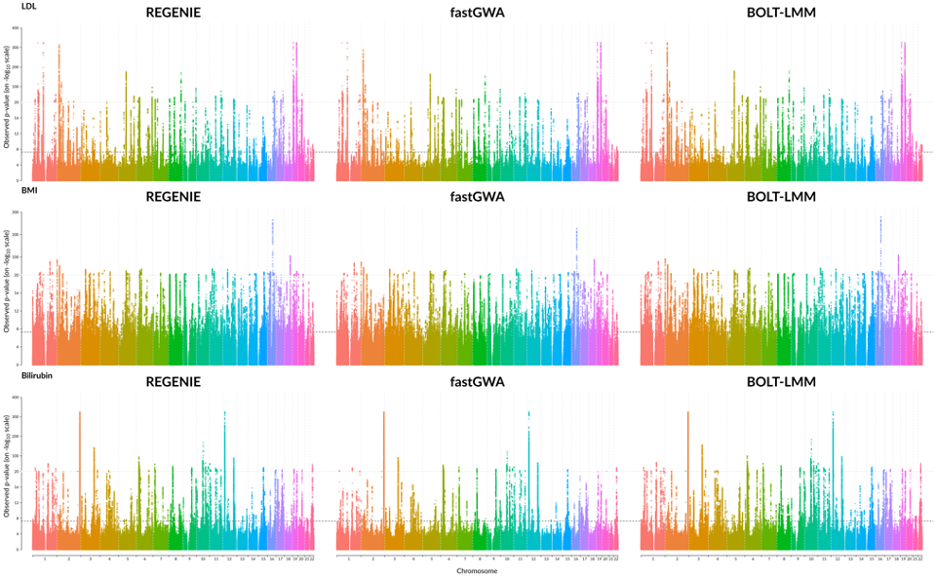
Fig. 1 Comparison of GWAS results for three quantitative phenotypes from the UK Biobank project, produced by REGENIE, BOLT-LMM, and fastGWA.
In addition to performance considerations, the whole genome regression approach produces covariates that are compatible with standard GWAS methods, and which eliminate spurious associations caused by population structure that are seen with traditional approaches. The Manhattan plots in figure 2 below compare the results of a traditional linear regression GWAS using standard covariates, to a linear regression GWAS using the covariates generated by WGR. This flexibility of GloWGR is another tremendous advantage over existing GWAS tools, and will allow for a wide variety of exciting extensions to the association testing framework that is already available in Glow.
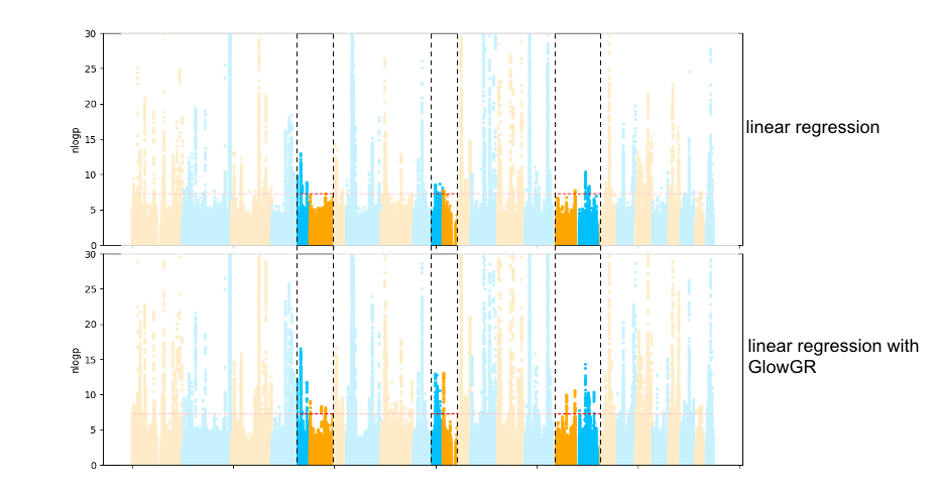
Fig. 2 Comparison of GWAS results of the quantitative phenotype bilirubin from the UK Biobank project, evaluated using standard linear regression and linear regression with GloWGR. The heightened peaks in the highlighted regions show the increase in power to detect subtler associations that is gained with GloWGR.
Figure 3 shows performance comparisons between GloWGR, REGENIE, BoltLMM, and fastGWA. We benchmarked the whole genome regression test implemented in Glow against the C++ implementation available in the single-node regenie tool to validate the accuracy of the method. We found that the two approaches achieve statistically identical results. We also found that the Apache Spark™ based implementation in Glow scales linearly with the number of nodes used.
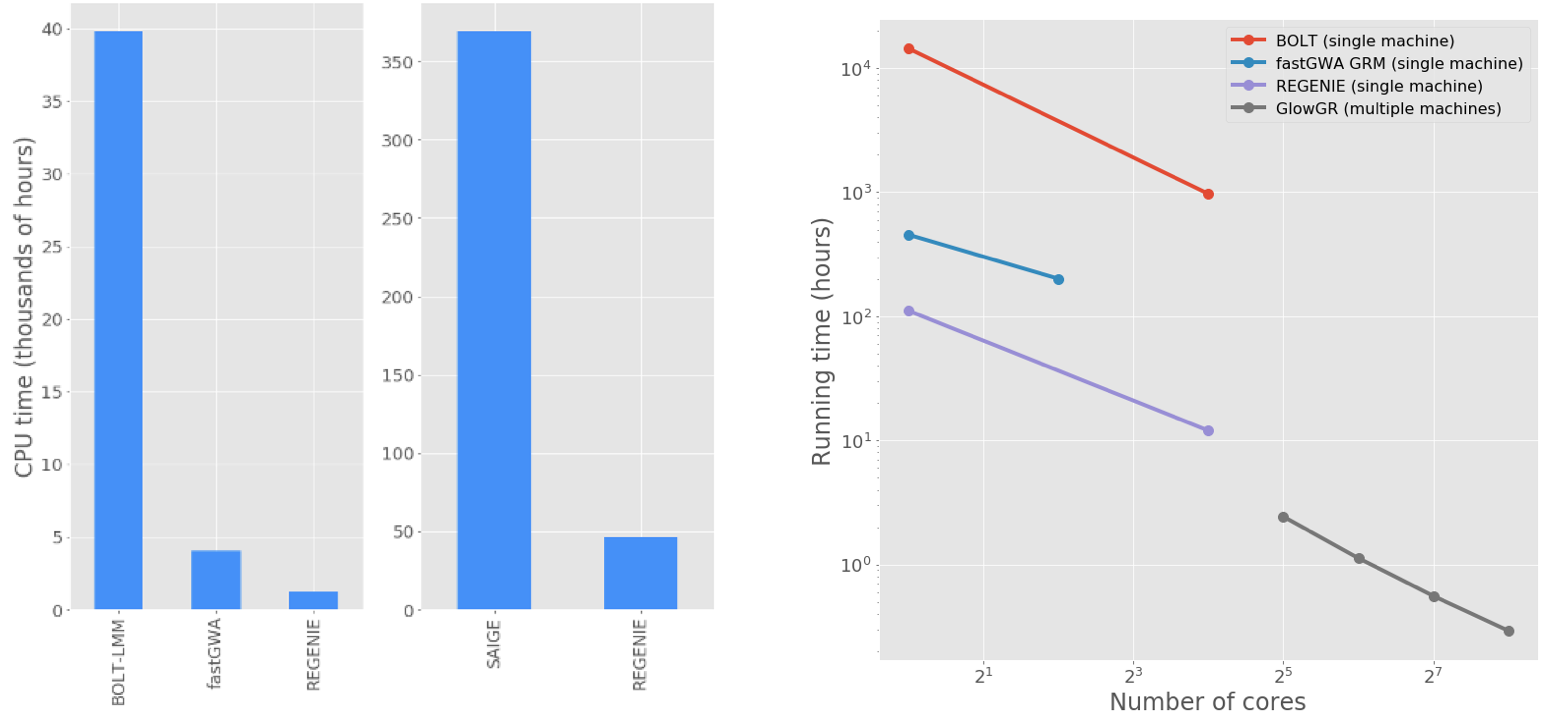
Fig. 3 Left: end-to-end GWAS runtime comparison for 50 quantitative traits from the UK Biobank project. Right: Run time comparison to fit WGR models against 50 quantitative phenotypes from the UK Biobank project. GloWGR scales well with cluster size, allowing for modeling of dozens of phenotypes in minutes without costing additional CPU efficiency. The exact list of phenotypes and computation environment details can be found here.
Scaling Whole Genome Regression within Project Glow
Performing WGR analysis with GloWGR has 5 steps:
Dividing the genotype matrix into contiguous blocks of SNPs (~1000 SNPs per block, referred to as loci)
Fitting multiple ridge models (~10) with varying ridge penalties within each locus
Using the resulting ridge models to reduce the locus from a matrix of 1,000 features to 10 features (each feature is the prediction of one of the ridge models)
Pooling the resulting features of all loci into a new reduced feature matrix X (N individuals by L loci x J ridge models per locus)
Fitting a final regularized model from X for the genome-wide contribution to phenotype Y.
Glow provides the easy-to-use abstractions shown in figure 4 for transforming large genotype matrices into the blocked matrix (below, left) and then fitting the whole genome regression model (below, right). These can be applied to data loaded in any of the genotype file formats that Glow understands, including VCF, Plink, and BGEN formats, as well as genotype data stored in Apache Spark™ native file formats like Delta Lake.

Fig. 4 Creating a matrix grouped by locus and fitting mixed ridge regression models using GloWGR
Glow provides an implementation of the WGR method for quantitative traits, and a binary trait variant is in progress. The covariate-adjusted phenotype created by GloWGR can be written out as an Apache Parquet ™ or Delta Lake dataset, which can easily be loaded by and analyzed within Apache Spark, pandas, and other tools. Ultimately, using the covariates computed with WGR in a genome-wide association study is as simple as running the command shown in Figure 5, below. This command is run by Apache Spark™, in parallel, across all of the genetic markers under test.

Fig. 5 Updating phenotypes with the WGR results and running a GWAS using the built-in association test methods from Glow
Join us and try whole genome regression in Glow!
Whole genome regression is available in Glow, which is an open source project hosted on Github, with an Apache 2 license. You can get started with this notebook that shows how to use GloWGR on data from 1,000 Genomes, by reading the preprint, by reading our project docs, or you can create a fork of the repository to start contributing code today.